
HEMOGRAMA

Es un examen de sangre que sirve para diagnosticar la anemia. Este consiste en un estudio cuantitativo y cualitativo de las tres series sanguíneas: Eritrocitos o Glóbulos Rojos, Leucocitos o Glóbulos Blancos y las Plaquetas. Para esto se incluyen varios parámetros cuantitativos, como el Hematocrito, la determinación de Hemoglobina, Recuento de Glóbulos Rojos, cálculo de las constantes Hematimétricas, como el Volumen Corpuscular Medio (VCM), Concentración de Hemoglobina Corpuscular Media (CHCM), Hemoglobina Corpuscular Media (HCM), RDW. Recuento de Reticulocitos; para evaluar cuantitativamente a la serie roja.
LINFOMA

Los linfomas son un conjunto de enfermedades cancerosas que se desarrollan en el sistema linfático, que también forman parte del sistema inmunológico del cuerpo humano. A los linfomas también se les llama los tumores sólidos hematológicos para diferenciarlos de las leucemias
CUANTOS TIPOS HAY
La principal clasificación de los linformas los divide en dos tipos según su origen celular, evolución, tratamiento y pronóstico que son:
La enfermedad de Hodgkin

También llamada linfoma de Hodgkin, es un cáncer que se origina en el tejido linfático. Este tejido comprende los ganglios linfáticos y los órganos relacionados que forman parte del sistema inmunológico y del sistema productor de sangre del cuerpo. Los ganglios linfáticos son órganos pequeños en forma de frijol que se encuentran debajo de la piel en el cuello, las axilas y la ingle. También se encuentran en muchas otras partes del cuerpo, por ejemplo dentro del tórax, el abdomen y la pelvis.
Los ganglios linfáticos producen y almacenan un tipo de glóbulos blancos, llamados linfocitos, encargados de combatir las infecciones y se comunican a través de todo el cuerpo mediante los vasos linfáticos (conductos estrechos similares a los vasos sanguíneos). Estos vasos linfáticos transportan un líquido acuoso e incoloro (líquido linfático) que contiene los linfocitos. Finalmente, el líquido linfático pasa a las venas localizadas en la parte superior del tórax.
Entre otros componentes del sistema linfático se encuentran el bazo, la médula ósea y el timo. El bazo es un órgano situado en la parte superior izquierda del abdomen y está compuesto principalmente de linfocitos maduros e inmaduros. Su función consiste en eliminar las células viejas y otras sustancias de desecho de la sangre. La médula ósea es el tejido esponjoso situado dentro de los huesos que crea nuevos glóbulos rojos y blancos, incluyendo los linfocitos. El timo es un pequeño órgano situado en el tórax que desempeña una función importante en la maduración de un linfocito especial llamado célula T
El linfoma no-Hodgkin (LNH)

Es un tipo de cáncer que surge en los linfocitos, un tipo de glóbulo blanco de la sangre. Se denomina de este modo para distinguirlo de la enfermedad de Hodgkin, un subtipo particular de linfoma. De hecho, es un término que incluye muchas formas diferentes de linfoma, cada uno con sus características individuales.
Los linfomas constituyen nódulos que se pueden desarrollar en cualquier órgano. La mayoría de los casos empiezan con una infiltración en un ganglio linfático (nodal), pero subtipos específicos pueden estar restringidos a la piel, cerebro, bazo, corazón, riñón u otros órganos (extranodal). El diagnóstico del linfoma requiere una biopsia del tejido afectado. El tratamiento del linfoma de bajo grado puede ser defensivo, pero el linfoma no-Hodgkin de alto grado se trata normalmente con quimioterapia y a menudo con radioterapia.
CUAL ES SU DIAGNOSTICO
Hodgkin
Biopsia
Hemograma
Velocidad de sedimentacion Globular
Bioquimica Sanguinea
Radiografia de Torax
Tomografia Axial Computarizada
No Hodgkin
Rayos x
Tomografia Axial Computarizada
Biopsia
Resonancia magnetica
LA ANEMIA

La anemia es un trastorno frecuente de la sangre que ocurre cuando la cantidad de glóbulos rojos es menor que lo normal, o cuando la concentración de hemoglobina en sangre es baja.
ANEMIA HEMOLITICA

La anemia hemolítica es un trastorno en el cual los glóbulos rojos se destruyen más rápido de lo que la médula ósea puede producirlos. El término para la destrucción de los glóbulos rojos es "hemólisis".
MANIFESTACIONES CLÍNICAS:

Anemia, esplenomegalia e ictericia (ictericia hemolítica congénita) que obedece al aumento de la concentración de bilirrubina no conjugada en el plasma. Es frecuente la litiasis por cálculos pigmentarios, incluso en la niñez. En los huesos largos se observa hiperplasia eritroide compensadora de la médula ósea acompañada de expansión de la médula hacia el centro de la diáfisis y, en ocasiones, de eritropoyesis extramedular, lo que a veces da lugar a la formación de masas paravertebrales visibles en la radiografía de tórax. Como la capacidad de la médula ósea para aumentar la eritropoyesis.
POLICITEMIA

La policitemia es un trastorno en el cual hay demasiados glóbulos rojos en la circulación sanguínea. Es el opuesto de la anemia, que ocurre cuando hay escasez de glóbulos rojos en la circulación. La policitemia también se denomina plétora (aumento excesivo de sangre).Esto puede ocurrir si a una dada presión de oxígeno la Hb queda saturada de O2 no liberándolo como las Hb normales en las mismas condiciones. El organismo para compensar origina más glóbulos rojos

La policitemia leve puede no causar problemas. Sin embargo, demasiados glóbulos rojos pueden aumentar el volumen o espesar la sangre, dificultando su circulación por el sistema sanguíneo y los órganos. Los bebés pueden tener dificultad para respirar y sus corazones y vasos sanguíneos no pueden compensar la cantidad adicional de sangre. A medida que las grandes cantidades de glóbulos comienzan a destruirse, se produce una sustancia llamada bilirrubina. El nivel alto de bilirrubina, llamado hiperbilirrubinemia, puede provocar ictericia, la coloración amarillenta de la piel, los ojos y las membranas mucosas. En recién nacidos, la hiperbilirrubinemia puede ocasionar kernicterus, una enfermedad por depósito de bilirrubina en el sistema nervioso. La policitemia también puede provocar convulsiones.
Síntomas

Muchos bebés con policitemia no tienen síntomas visibles del trastorno. A continuación se enumeran los síntomas más comunes de la policitemia. Sin embargo, cada bebé puede experimentarlos de una forma diferente. Los síntomas pueden incluir:

• coloración rojiza-púrpura profunda
• mala alimentación bajo nivel de glucosa en sangre
• A veces dolor de cabeza por congestión de vasos.
• Rotura de vasos.
• Trastornos de circulación.
• Trombosis.
Diagnóstico
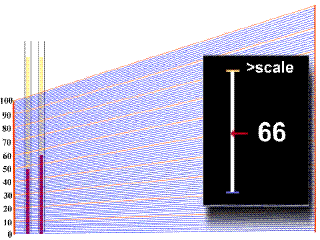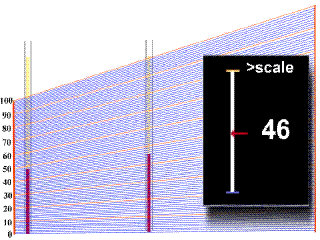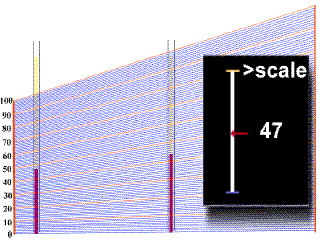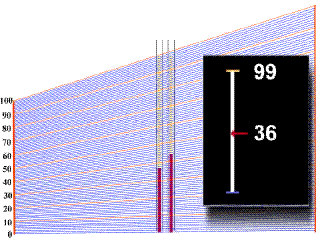
Tratamiento

El tratamiento puede incluir:
• Extraer una parte del volumen de sangre, reduciendo así la cantidad de glóbulos rojos.
• Reemplazar la sangre extraída con líquidos (para ayudar a diluir la concentración de glóbulos rojos).
• Exsanguíneo transfusión parcial (extraer y reemplazar lentamente una gran parte del volumen de sangre del bebé).
Estos tratamientos se llevan a cabo a través de una vena o arteria, a menudo de los vasos sanguíneos umbilicales
Tipos
Policitemia se refiere al aumento del número de hematíes y, generalmente, de la cantidad de hemoglobina. Hay dos tipos:
• Policitemia esencial o Policitemia vera:

Es de causa desconocida, aunque muchas veces está en el contexto de un síndrome mieloproliferativo.
• Policitemia secundaria a:

- Algunos tipos de tumores.
- Endocrina: en el síndrome de Cushing.
- La compensadora, en situaciones en que la médula ósea, si hay poco oxígeno, fabrica más hematíes, como por ejemplo en patologías de pulmón
ERITREMIA

Eritemia crónica: una enfermedad de la sangre caracterizada por la presencia de un exceso de eritrocitos.
Concepto

La policitemia vera, también llamada policitemia primaria, eritremia, policitemia rubra vera o enfermedad de Vázquez - Osler es un síndrome mieloproliferativo crónico en el cual ocurre un incremento de las células sanguíneas, principalmente de los hematíes. No obstante, también suele presentar leucocitosis y trombocitosis. Afecta principalmente a varones, en edades comprendidas entre los 50 y los 60 años. Es una enfermedad de inicio insidioso y desarrollo lento.
Sintomas

• Síntomas derivados de hiperviscosidad sanguínea: el aumento de la masa celular de la sangre y el consiguiente enlentecimiento circulatorio puede producir, entre otros:
o Coloración rojiza de la piel, especialmente en la parte superior del cuerpo. El enfermo presenta un rostro enrojecido clásicamente denominado facies eritroide.
o Conjuntivitis bilateral.
• Repercusiones hemodinámicas de la hipoxia miocárdica.
o Disnea.
o Hipertensión arterial.
o Ángor
• Fenómenos trombóticos y hemorrágicos, derivados de la hiperviscosidad sanguínea y alteraciones de la hemostasia; son más frecuentes y graves en el cerebro.
Historia clínica: anamnesis y exploración. Importante realizar un examen de fondo de ojo (dilatación venosa, hemorragias retinianas...).
2. Pruebas analíticas:
• Hematíes > 5,5 millones/ml, con aumento de la masa eritrocitaria > 36 ml / kg en varones y > 32 ml / kg en mujeres.
• Hemoglobina > 19 g / dl en varones y > 18 g / dl en mujeres.
• Hematocrito > 55% en varones y > 50% en mujeres.
• Leucocitosis y trombocitosis.

• Disminución de la velocidad de sedimentación globular (valores de 0,1 - 0,2 mm).
• Bioquímica con eritropoyetina e hierro normales.
1. Biopsia de médula ósea: hipercelularidad de las tres líneas.
2. Pruebas de imagen: para hacer diagnóstico diferencial con patologías causantes de policitemia secundaria (ecografía de abdomen, de ovarios, radiografía de tórax...).
Criterios diagnósticos
Deben cumplirse los tres mayores o los dos primeros mayores y dos menores.
Criterios mayores.

• Aumento de la masa eritrocitaria.
Varon: más de 36ml/kg
Mujer: más de 32ml/kg
• Saturación oxigeno > 92%.
• Esplenomegalia.
Criterios menores

• Trombocitosis.
• Leucocitosis.
• Aumento de la fosfatasa alcalina granulocítica.
• Ausencia de fiebre e infección.
• Vitamina B12 > 90 ηg / ml.
Complicaciones
Puede evolucionar a leucemia mieloblástica aguda o a mielofibrosis con metaplasia mieloide.
Tratamiento

Tratamiento crónico, de un mínimo de 6 a 8 años. Consiste en:
1. Flebotomías para corregir la hiperviscosidad y la clínica asociada, a razón de 500 ml / semana durante 4 - 6 semanas.
2. Quimioterapia con hidroxiurea vía oral, para reducir la proliferación eritrocitaria.
3. Antiagregantes plaquetarios, como el ácido acetilsalicílico (AAS) para evitar los fenómenos trombóticos.
4. Colestiramina o baños con sales si existe prurito.
LEUCEMIA

La leucemia o leucosis es un grupo de enfermedades malignas de la médula ósea (cáncer hematológico1 ) que provoca un aumento incontrolado de leucocitos (glóbulos blancos) clonales en la médula ósea, que suelen pasar a la sangre periférica aunque en ocasiones no lo hacen (leucemias aleucémicas). Ciertas proliferaciones malignas de glóbulos rojos se incluyen entre las leucemias (eritroleucemia).
TALASEMIA

Talasemia (del griego θάλασσα, thalassa, que significa mar) es un grupo de enfermedades hereditarias1 en las que se produce un defecto en la síntesis de la hemoglobina. El nombre es debido a que esta enfermedad fue primeramente descrita en poblaciones costeras al Mediterráneo; sin embargo, la enfermedad también es prevalente en África, Oriente medio y Asia.2
Síntomas
El defecto o deleción de un gen en la talasemia β causa una anemia hemolítica que oscila entre leve y moderada sin síntoma alguno. La deleción de dos genes ocasionan anemia más severa y la presencia de síntomas: debilidad, fatiga, dificultad respiratoria. En las variantes más graves, como la talasemia beta mayor, pueden aparecer ictericia, úlceras cutáneas, cálculos biliares y agrandamiento del bazo (que en ocasiones llega a ser enorme). La actividad excesiva de la médula ósea puede causar el ensanchamiento y el agrandamiento de algunos huesos, especialmente los de la cabeza y del rostro.

Los huesos largos tienden a debilitarse y fracturarse con gran facilidad. Los niños que padecen ciertas talasemias pueden crecer con más lentitud y llegar a la pubertad más tarde de lo normal. Como la absorción del hierro puede aumentar como respuesta a la anemia sumado al requerimiento de transfusiones de sangre frecuentes (las cuales suministran más hierro), es posible que se acumulen cantidades excesivas de hierro y se depositen en la musculatura del corazón, causando insuficiencia cardíaca.
Clasificación

• α Talasemia rasgo (portador). Las mutaciones de la cadena α en el cromosoma 16 afecta a uno de los genes de un cromosoma causando una talasemia silenciosa (asintomática) caracterizada por algunas hemoglobinas con tres β y una α globina. Los portadores pueden tener mutaciones de la cadena α en dos cromosomas (16) afectando a un genes de cada cromosoma (o bien los dos genes de un solo cromosoma, estando normales los dos del cromosoma homólogo) causando una talasemia leve (puede ser asintomática) caracterizada por hemoglobinas con tres β y una α globina. Debido a que existen suficientes genes sin mutación en la cadena α la mayoría de las moléculas de hemoglobina tienen las respectivas dos cadenas α y dos β. La mayoría de los portadores de la α talasemia no lo saben y se descubre con análisis de ADN y biología molecular.

• α Talasemia grave (Hemoglobina H). Las mutaciones de la cadena α en el cromosoma 16 afecta a tres de los genes (involucrando a ambos cromosomas homólogos) causando una talasemia grave caracterizada por la mayoría de las hemoglobinas con tres cadenas β y una α globina. Los afectados cursan con hemolisis intravascular causando anemia severa con los síntomas más graves.

• α Talasemia Mayor (Enfermedad de Bart). Las mutaciones de la cadena α en el cromosoma 16 afecta a los cuatro genes (involucrando a ambos cromosomas homólogos) causando un hidrops fetal caracterizada por hemoglobinas con solamente cuatro cadenas γ (gamma) y es incompatible con la vida.

• β+ Talasemia Menor (Minor). Las mutaciones de la cadena β en el cromosoma 11 afecta a uno de los genes causando una talasemia relativamente leve caracterizada por una hemoglobina con tres α y una β globina. Puede que no haya síntomas como puede que los síntomas sean intermedios entre leve y graves.
+o+Anemia+de+Cooley.jpg)
• βº Talasemia Mayor (Major) o Anemia de Cooley.7 Las mutaciones de la cadena β en el cromosoma 11 afectan a ambos genes causando la más grave de las talasemias caracterizada por la falta total de β globina. Cuatro cadenas α se combinan en defecto de las cadenas β formando una hemoglobina inestable que tiende a precipitarse en los glóbulos rojos causando daños en la membrana celular e incrementando la fragilidad del hematíe en cuestión. Por razón de la masiva hemolisis dirigida por el bazo, los síntomas son más graves: palidez, suceptibilidad a infecciones, fragilidad ósea, ictericia, depósitos de hierro en el hígado y corazón, y puede que no vivan mucho tiempo. El tratamiento consiste en transfusiones sanguínea



